Lexin Chen
About

Welcome!
My path into computational science began with a fascination for the elegant machinery of proteins like ATP synthase, nature's own rotary engine. This sparked a central question: how can we use computation to understand and simulate this incredible complexity? I am now dedicated to breaking down the computational bottlenecks that prevent scientists from learning from their largest and noisiest datasets.
As a Computational Scientist, I combine machine learning and high-performance software engineering to build robust tools for drug discovery. I have engineered scalable analysis pipelines for molecular libraries and developed ML frameworks that perform consistently on real-world data, transforming complex research challenges into deployable solutions that accelerate progress.
Projects
DELight
Faced with the critical bottleneck of extreme class imbalance in billion-compound DNA-Encoded Libraries, where traditional ML models failed, I developed the DELight framework. I designed and benchmarked novel, targeted undersampling strategies to selectively curate training data. The result was a robust solution that significantly improved model generalizability and reliable hit identification, transforming massive, skewed datasets into actionable discovery tools.
Technologies used: Python
MDANCE
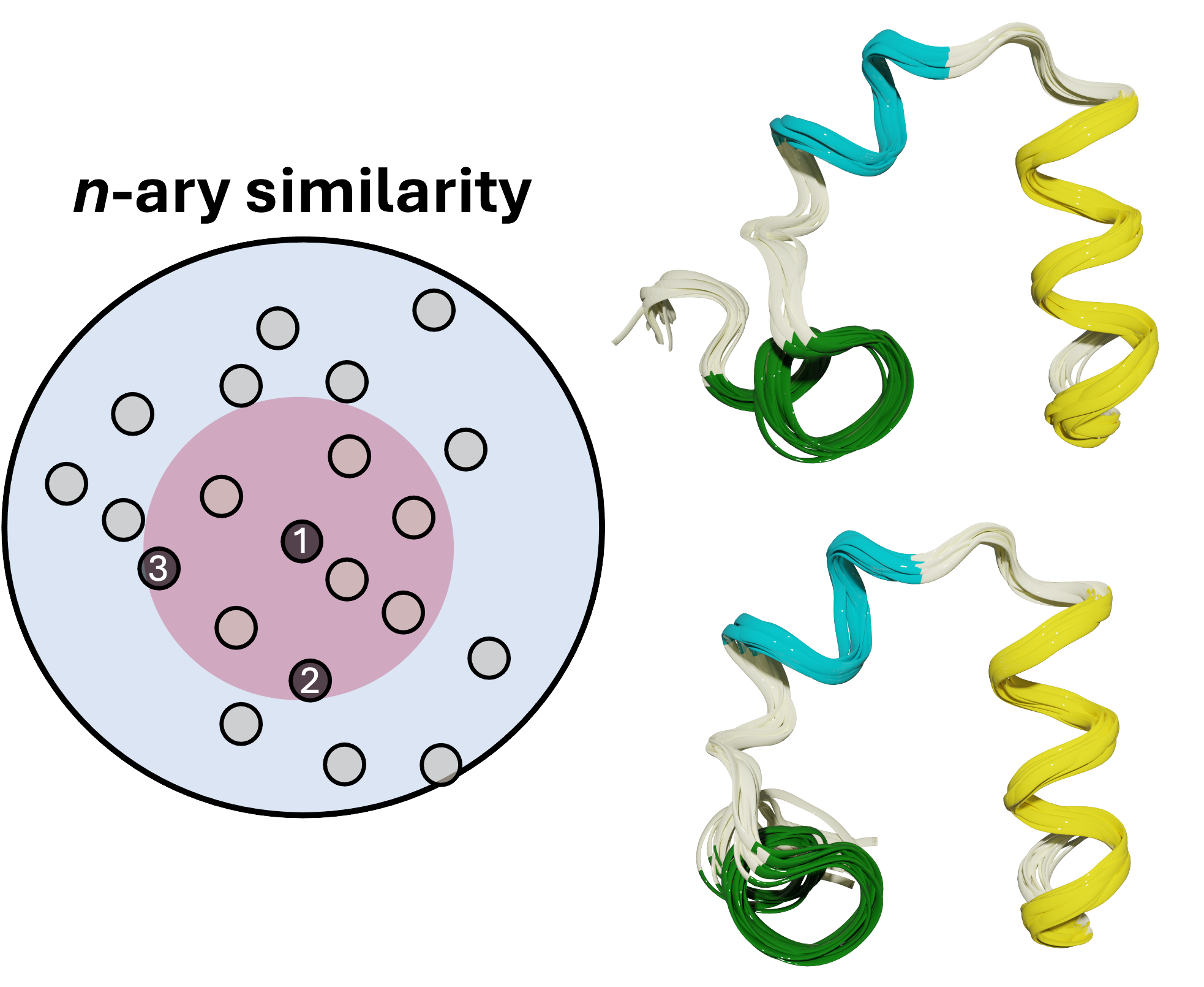
Conventionally, the computational bottleneck of analyzing massive molecular datasets and ultra-long simulations using traditional clustering methods was prohibitively slow. My solution was to pioneer a novel, linear-time clustering algorithm, architecting the MDANCE framework to be inherently modular and scalable by design. The result was a groundbreaking 25x speedup, enabling efficient similarity search and pattern recognition in million-scale molecular libraries and making large-scale analysis feasible for the first time.
Technologies used: Python
PRIME
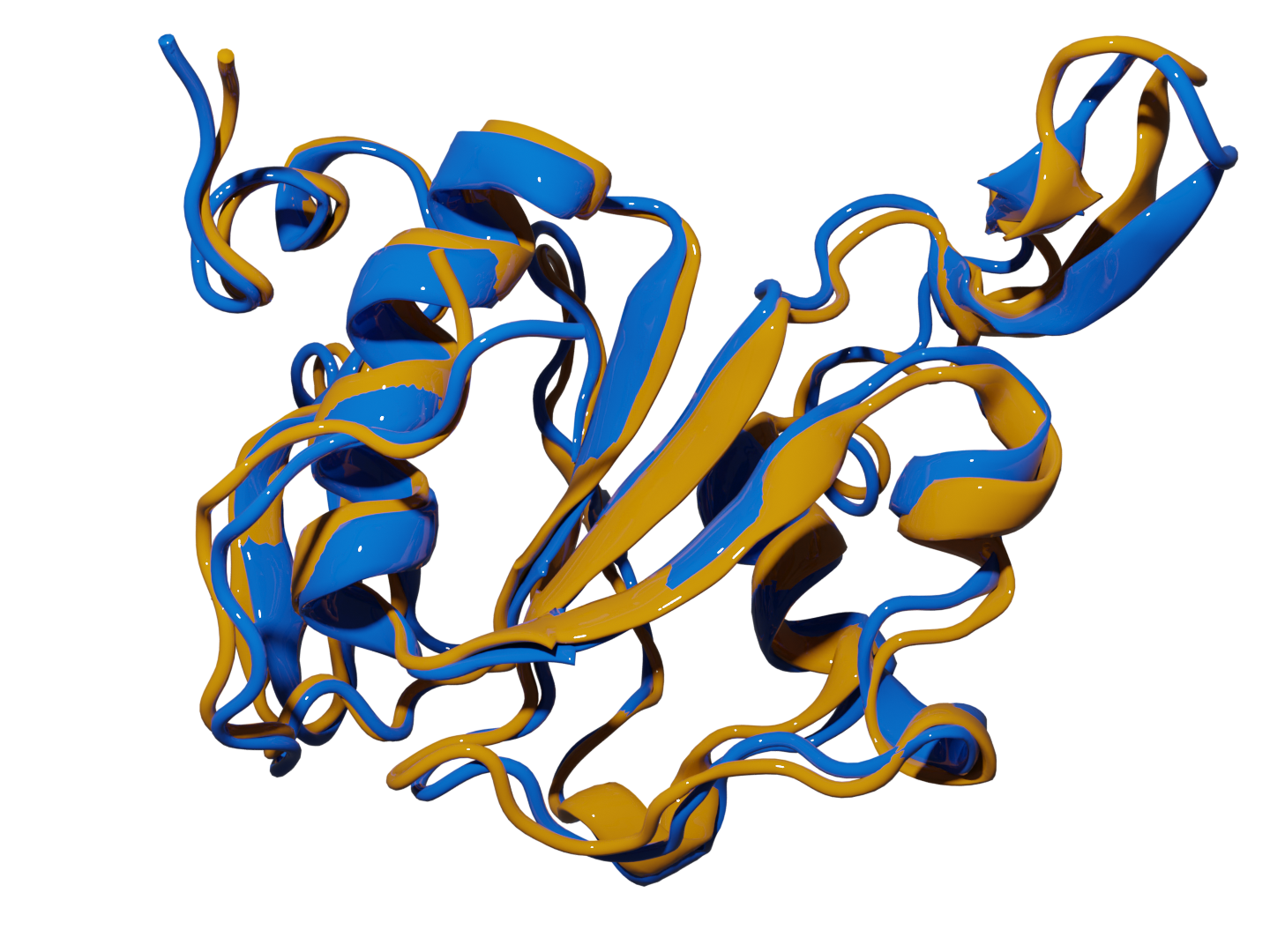
Accurately identifying a protein's true native structure from thousands of simulated conformations is a major bottleneck in structural biology, directly impacting our ability to understand function and design drugs. To solve this, I developed PRIME (Protein Retrieval via Integrative Ensembles), a novel tool that uses extended continuous similarity to intelligently pinpoint the definitive structure from complex ensembles. The result was a breakthrough in accuracy and efficiency: PRIME perfectly mapped all structural motifs across diverse challenging systems, achieving this with unprecedented linear scaling to make high-fidelity structural refinement both reliable and scalable.
Technologies used: Python
Quantifying Specific Ions Effect
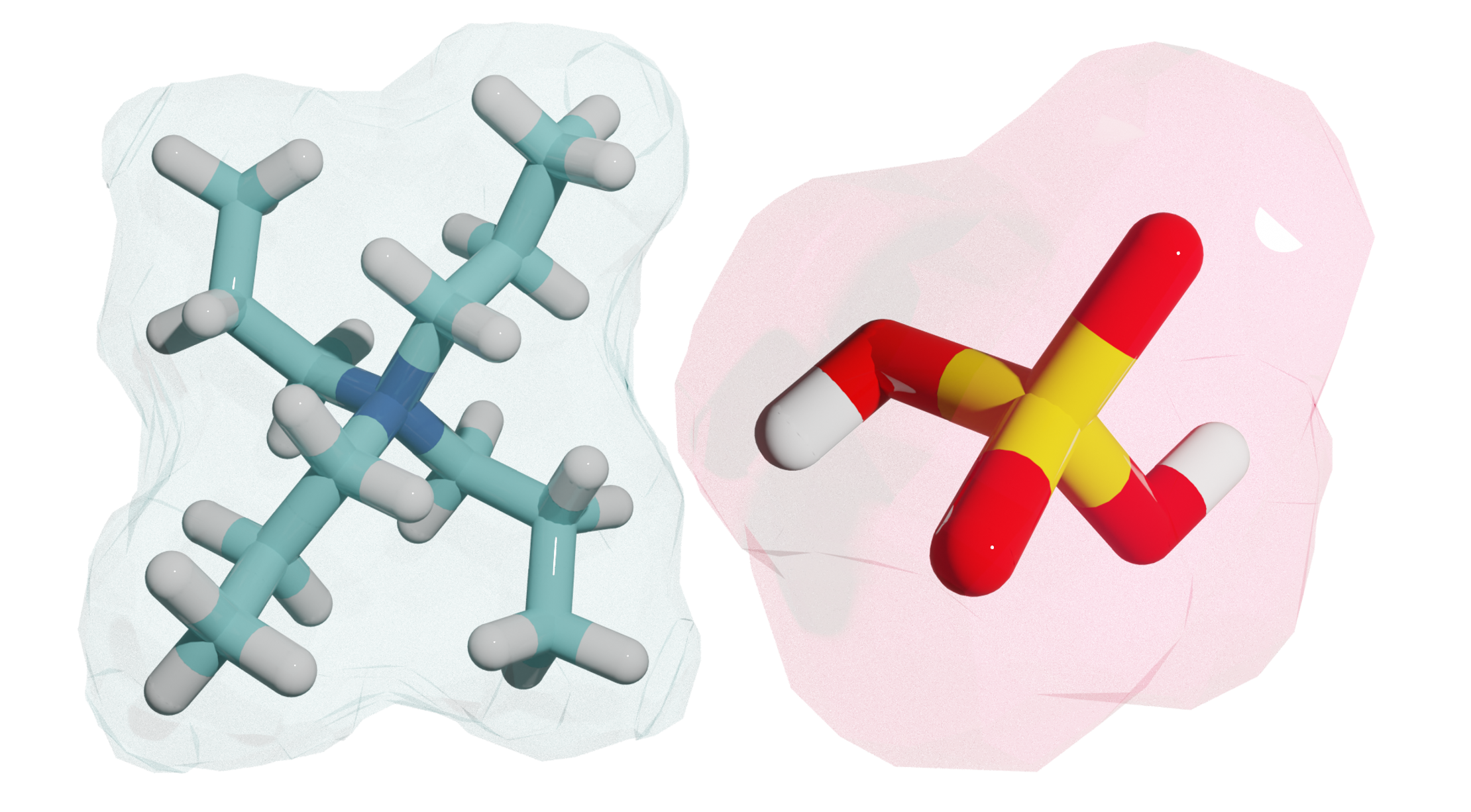
The mystery of ion selectivity in electrolyte solutions has puzzled scientists for centuries, with little known about why nature prefers one ion over another, a phenomenon known as the specific ion effect. I tackled this challenge by developing a computational framework that moves from qualitative observation to quantitative prediction. Using conceptual Density Functional Theory (DFT), I created models that capture the essential electronic properties of ions to accurately forecast their behavior. This work provides a principled, physics-based method to finally decipher the rules behind nature's selective preferences.
Technologies used: Python
MD Starters
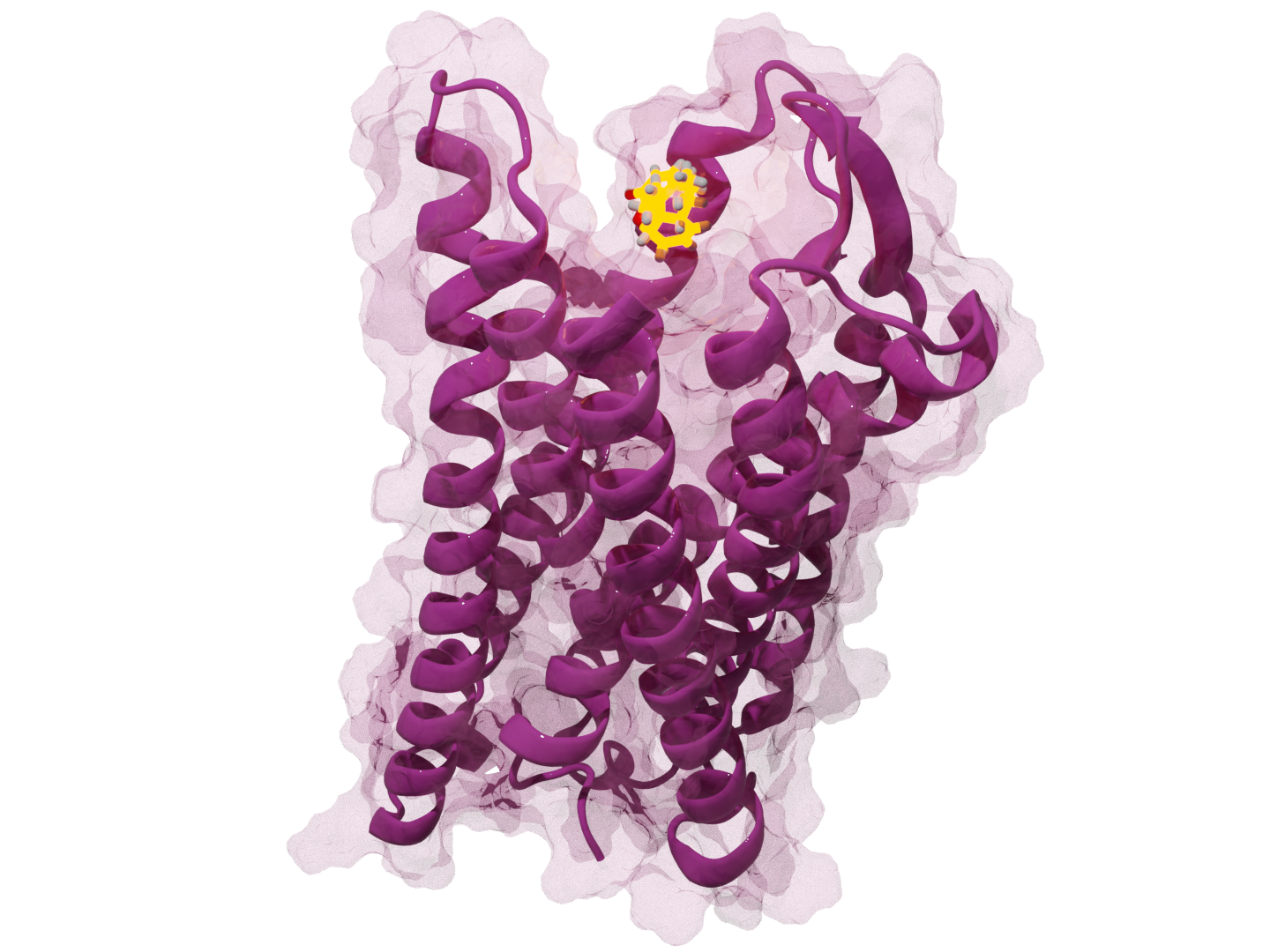
md-starters provides sample code for preparing and analyzing molecular dynamics simulations, mainly for membrane proteins.
Technologies used: Python
Technical Skills
Python
80%
R
40%
C++
20%
Bash Script
60%
HTML
40%