Lexin Chen
About

Machine Learning for Drug Discovery
My path into computational science began with a fascination for the elegant machinery of proteins like ATP synthase, nature's own rotary engine. This sparked a central question: how can we use computation to understand and simulate this incredible complexity? I am now dedicated to breaking down the computational bottlenecks that prevent scientists from learning from their largest and noisiest datasets.
As a Computational Scientist, I combine machine learning and high-performance software engineering to build robust tools for drug discovery. I have engineered scalable analysis pipelines for molecular libraries and developed ML frameworks that perform consistently on real-world data, transforming complex research challenges into deployable solutions that accelerate progress.
Projects
DELight: ML for DEL screening
Problem: DNA-encoded libraries (DEL) are extremely imbalanced, causing standard machine learning models to miss true binders. Built: Targeted undersampling framework to rebalance training data and improve signal detection. Impact: Increased generalization and hit identification by 5-10% on million-scale compound libraries.
MDANCE: Scalable Clustering for Molecular Simulations
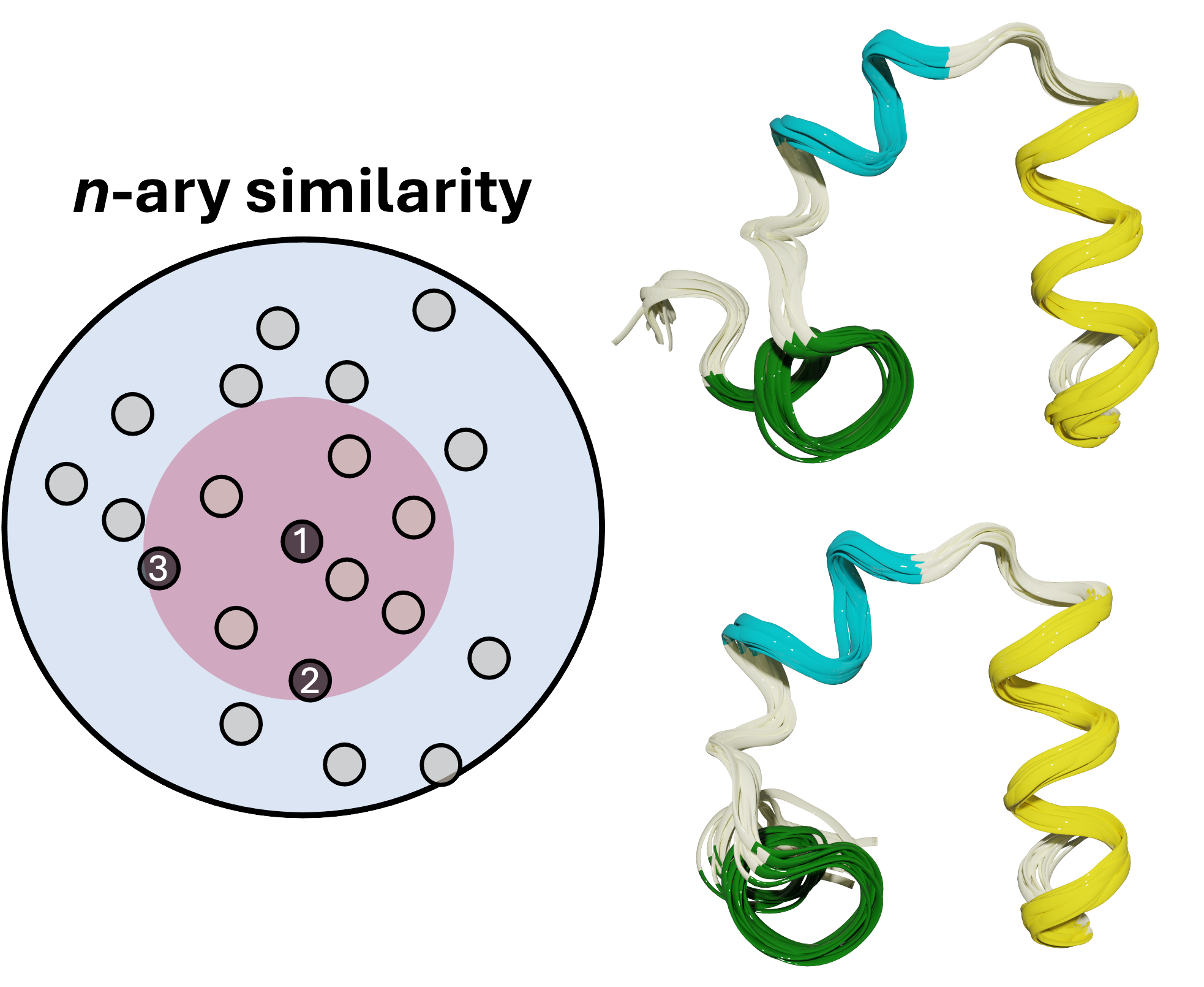
Problem: Clustering million-scale Molecular Dynamics trajectories is too slow. Built: Linear-time clustering algorithm and optimized implementation. Impact: 25x speedup on 1.5 million frames, enabling practical large-scale analysis.
PRIME: Native Structure Determination
Problem: Accurate protein structural retrieval prediction is limited by data scale and model efficiency. Built: An algorithm for representative structure selection to identify key structures from molecular dynamics ensembles. Impact: Achieved perfect recall of critical conformational states with high computational efficiency, enabling rapid analysis for docking and virtual screening pipelines.
Technical Skills
Python
80%
R
40%
C++
20%
Bash Script
60%
HTML
40%